It has been a year since the more stringent Medical Device Regulation (MDR) replaced the Medical Device Directive (MDD). We are currently in a transition period, and slowly but steadily we see the number of products that comply with the MDR increase. But with two years left on the clock, only 17% of the AI products for radiology have obtained clearance under the MDR. If the market continues at this pace, half of the currently CE marked AI products for radiology will not comply in time. The likely consequence is that software will be taken off the market and can no longer be used clinically after May 2024.
Should users stop buying MDD compliant AI software? Probably not just yet. Is it worth considering the regulatory status and asking some critical questions when choosing a new product? That’s not a bad idea.
Before we continue, let’s refresh your memory and explain a few of these abbreviations from the wonderful world of regulatory compliance. If you are a regulatory aficionado - feel free to skip the next bit.
MDD, MDR, CE, what are you talking about?
What I am talking about here is regulatory clearance. It is what differentiates a research or prototype product from a product that may be used in clinical practice for patient care. In the United States, the Food and Drug Administration (FDA) provides clearance for medical devices, including AI software (yes, software is considered a medical ‘device’ as well). In Europe the European Commission develops rules to ensure that medical devices entering the market are safe. Vendors are audited by organizations called Notified Bodies who check conformity to these rules. If a Notified Body gives its stamp of approval, a manufacturer can affix a CE mark to their product. A CE mark indicates that a medical device complies to the European rules and allows it to enter the market for clinical use.
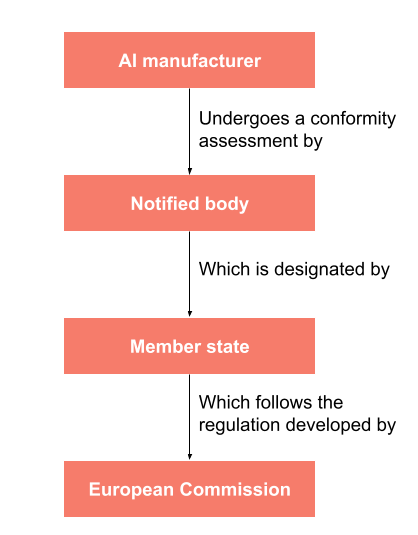
Parties involved in ensuring medical devices (including AI software) are safe and effective for use in the European Union.
In 2021, the Medical Device Directive (MDD), was replaced by the Medical Device Regulation (MDR). The MDD came out in 1993 and was well due for an update. Scandals such as the PIP breast implants highlighted that the directives were no longer fit for purpose. Moreover, the directives provided little in the way of guidance for more modern devices including those that use software. In 2017 the MDR was ratified. One year ago, on May 26th 2021, the MDR came into effect.
This changed
So what changed? Too much to discuss in detail here, but the two most notable changes are:
-- The former MDD allowed for some medical AI software tools to be classified as a low-risk, class I device. Vendors could self-certify their products in this case. No external audit by a Notified Body was needed. However, with the MDR, AI software for radiology will mostly be classified as a class IIa or IIb device, due to new classification rules, which means an audit from a Notified Body is always needed.
-- The new regulations put more emphasis on demonstrating clinical efficacy and performing post-market surveillance. Vendors will have to actively monitor the functioning and impact of their software in the field.
All in all, the idea is that the MDR will improve the safety and performance of medical (AI) devices on the European market. Check out Emilia Niemiec’s paper: “Will the EU Medical Device Regulation help to improve the safety and performance of medical AI devices?” for the extensive explanation.
For the regulatory aficionados, pick it back up here.
The consequences
Practically, the change from MDD to MDR means that all AI software currently on the market needs to be recertified under the MDR. We are now in a transition phase to give vendors the time to get this done. The products already on the market under the MDD may be clinically used until May 2024, but are constrained to minor updates only. Any new products or large updates need to be (re-)certified and conform to the MDR. All vendors, MDD or MDR, have to meet the requirement of post marketing surveillance, proactively gathering information on the real-world use and effectiveness of their products. Devices not meeting the requirements after May 2024 will have to withdraw from the market and can no longer be used in clinical practice. Unless this deadline is extended, something that happened before.
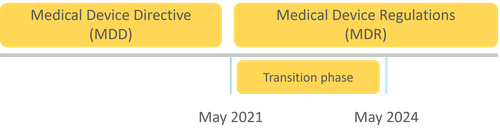
The MDD was replaced by the MDR in May 2021. Until 2024 (if not extended) there is a transition phase for existing medical device vendors to become compliant.
The regulatory landscape now
One year after the introduction of the MDR, only 33 products (17%) out of the 198 CE marked AI products listed on www.AIforRadiology.com comply with the MDR. A third of the products are on the market as a class I MDD device. All of those will be class IIa products under MDR and this means the vendors will have to do a serious amount of work to update their documentation and processes and gain approval from a Notified Body to stay on the market with their product after 2024. If we continue at this pace, only half of the AI products will make it before May 2024.
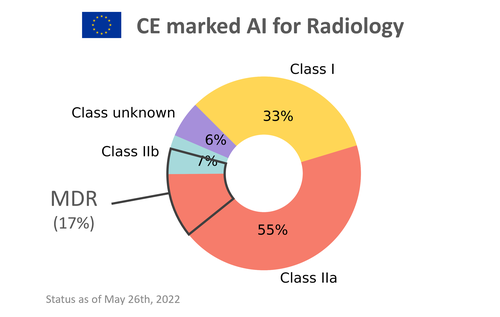
Risk class of CE marked AI for radiology software. 33 products (17%) are MDR compliant one year after the introduction of the MDR.
Looking at the radiology AI market development in recent years, I can’t help but notice a correlation between the stagnation of growth and the introduction of the MDR. In 2019 and 2020, many new products came to market (4-5 years after a flurry of companies were founded, mostly using the breakthrough technology of deep learning). In 2021 we observed stagnation. Two thirds of the new products that year have a certification date in the month of May. That is no coincidence as the MDR was introduced on the 26th of May. Many vendors pushed hard to get their product on the market as an MDD device just before that date, giving them another 3 years of slack.
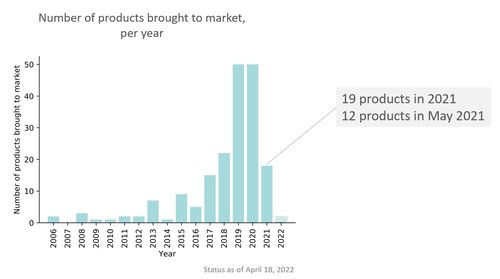
New CE marked AI for radiology software products per year. Majority of new products introduced in 2021 were brought to market just before the MDR deadline (26th of May 2021)
Also noticeable is that since the introduction of the MDR, only one new vendor has entered the field (Ligence, introduced to you last month). The other products were portfolio expansions of existing vendors. We may wonder if compliance with the more strict MDR is forming a major hurdle for new players to enter the field. A related issue is that also Notified Bodies need to implement changes to comply with the MDR, and several have not succeeded, so there are fewer Notified Bodies for companies to go to. Or is the market for AI products in radiology simply saturated and not attractive for newcomers anymore? I guess a combination of both may be the cause.
So should I not buy MDD products?
With all that said, what now? If you are a (future) user or buyer of AI software, this might trigger some alarm bells. How important is the CE status when choosing AI software to implement (or any other medical device in that respect)? Should you avoid purchasing any MDD complying products?
I believe it’s a bit more nuanced than that. Obviously for those products who comply with the MDR, you have a bit more certainty they will remain on the market in the coming years. For the MDD certified products, there is more risk, but it may not be a reason to ignore them all together. There are important reasons for vendors to not have CE marking under MDR just yet. 1) Recertification is not free, so there is a financial incentive to wait as long as possible. They may have been certified in recent years and want to utilise the full duration of their valid certification. 2) There is a shortage of Notified Bodies, which may mean a vendor is waiting in line to get MDR certified.
What to consider
The first to consider is the class of the product. Products with class IIa MDD have previously been audited by a Notified Body to be CE marked. This means they already have a contract with a Notified Body, have the most important processes in place, and have gone through the whole process before. Among Class I products, there may be more diversity between companies in how mature/ready/compliant they really are, as they have been self-certified. Here a prospective buyer may exercise some caution.
Secondly to consider is the roadmap. When talking to vendors with MDD compliance, it may be a good idea to ask them about their plans to comply with the MDR in the near future. Until when is their MDD certification valid? Do they have a contract with a Notified Body in place? When do they expect to be MDR compliant? Is there a dedicated RA/QA person on the team to make it happen? Is their funding guaranteed to cover the costs? In case the account manager has little clue of what you are talking about: this is not a good sign.
Lastly, think about what your purchasing contracts state. When deciding on contracts surpassing 2024, make sure to have some exceptions in place that you will not be paying in case the products do not meet the requirements needed at that point in time.
To conclude
Should users stop buying MDD compliant AI software? Probably not yet. Is it worth considering the regulatory status and asking some critical questions when choosing a new product? Not a bad idea.
Big thanks to medical software and regulatory experts Sandy Wright and James Dewar from Scarlet and prof. Bram van Ginneken for proofreading this blog.
